Ana Ion and Florinel Gabriel Banica
1) Department
of Analytical Chemistry and Instrumental Analysis, Politehnica
University of Bucharest , Bucharest, 1 Polizu Street, 78126,
Romania.
2) Department of Chemistry, Norwegian
University of Science and Technology (NTNU), 7491 Trondheim,
Norway.
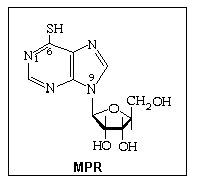
Key words: anticancer drugs, 6-mercaptopurine-9-D-riboside, polarography,
voltammetry, electrocatalysis, nickel complexes
Among anticancer drugs, antimetabolites form a distinct class. Their action consists in of the interference with the production of nucleic acid in tumor cells [1] . Sulfur derivatives of purine (6-mercaptopurine, MP, and thioguanine, TG) belong to this class and are especially employed in the treatment of acute leukemia in children [1-3]. The discovery of these drugs was rewarded by the Nobel Prize in 1988 [2] . An essential point is that the actual active compound under MP therapy not MP itself but its derivative, 6-mercaptopurine-9-D-riboside (MPR) that is produced by the metabolic transformation of MP. That is way direct administration of MPR appears as an alternative to theadministration of MP [1]. Consequently, analytical methods for MPR determination in either drug formulations or body fluids are highly required. Electroanalytical chemistry can provide advantageous solutions for both batch analysis and MPR detection in flowing fluids.
Although the well-known electrochemical reactivity of the MP fragment [4] may prove useful in this respect, the intricate reaction mechanism for direct reduction of this compound may be a serious drawback. That is why new possibilities are explored, especially those involving an indirect participation of the analyte in the electrode process. In this respect, nickel reduction catalyzed by MPR was advanced as a detection method in cathodic stripping voltammetry of the above compound [5]. At the same time the kinetics and the mechanism of Ni2+ reduction catalyzed by MPR was investigated by linear scan voltammetry [6]. Both above investigations were undergone by means of the hanging mercury drop electrode. Additional information on this topic, resulted from electrochemical investigations by means of the dropping mercury electrode, are presented in this repport.
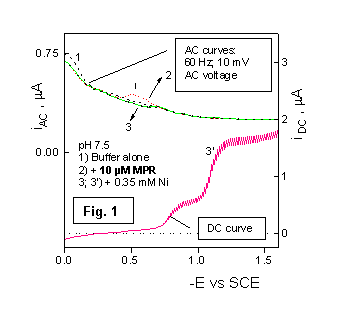
The occurrence of the catalytic nickel reduction in this system, at a MPR concentration of 10 µM, is demostrated by the first wave on curve 3 in Fig. 1, whereas the second wave on the same curve results from the direct reduction of the hydrated nickel ion. AC polarograms in Fig. 1 show no particular pattern, except for the potential range around 0.5 V, where the anodic reaction of mercury occurs, leading to the formation of a sparingly soluble complex with MPR. In agreement with the electrocapillary data [6], the AC polarograms in Fig. 1 demonstrate the absence of MPR adsorbtion in the potential range of the catalytic prewave.
The catalytic prewave is ascribed to an electrode process consisting of two steps according to the following Scheme, where L- stands for the ionized MPR molecule:
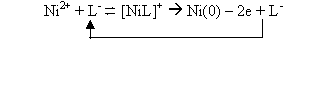
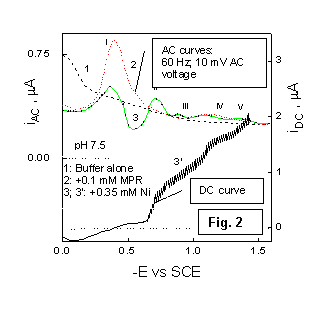
Substantial changes in the curve shape occur at higher MPR concentrations (0.1 mM), as shown in Fig. 2. On the DC polarogram, the catalytic nickel reduction produces a composite wave, whereas the AC polarogram recorded in the presence of both Ni2+ and MPR (curve 3) displays several peaks (labelled by II and III) in the region of the catalytic prewave. Under the same condition, electro-capillary curves demonstrate the occurrence of MPR adsorbtion [6].
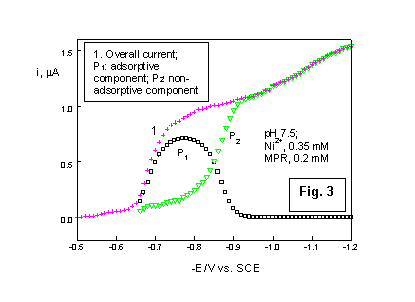
The above findings enable one to perform the interpretation of the prewave shape at higher concentrations by assuming the occurrence of two different mechanism for the catalytic nickel reduction. Each mechanism produces a distinct component of the overall current, as shown by curves P1 and P2 in Fig. 3. One of them (P2) involves non-adsorbed MPR as catalyst and the corresponding current-potential relationship is that characteristic for the irreversible polarographic wave, i.e.:
E = E1/2 + (RT/a nF) ln [i/(il i)]
Here, il is the limiting current of the P2 component and other symbols have the usual meaning.The above equation fairly describe the shape of the prewave recorded at a very low MPR concentration, where the adsorption effects are negligible (Fig. 1).
The second reaction path occurs with the participation of adsorbed MPR molecules. However, as demonstrated by electrocapillary data [6a], the adsorption degree is strongly dependent on the electrode potential in the region of the catalytic prewave. This enables one to represent the the current-potential relationship for the adsorptive component P2 (Fig. 3) by the equation derived for the
reduction of an adsorbed compound taking into account the effect of electrode potential on the constant in the Henry adsorption isotherm [7]:
According to the above interpretation, the shape of the nickel prewave
recorded at MPR concentrations over 10 µM results from the superposition
of the components P1 and P2. This makes difficult
to investigate the adsorptive component alone, whereas the non-adsorptive
component (P2) can reliabely be recorded at MPR concentrations
under 10 µM (with [Ni2+]/[MPR] > 30).
Under the above conditions, the catalytic prewave shows the typical
characteristics of the electrode processes with a chemical reaction as
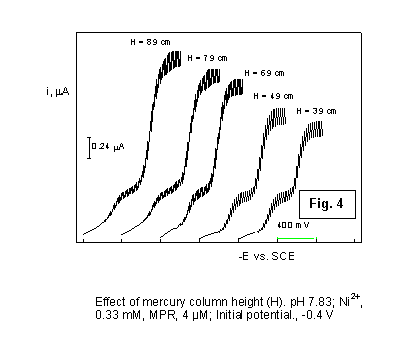
the rate-determining step. Thus, the prewave limiting current is not dependent
on mercury pressure above the lower end of the capillary electrode (Fig.
4), proving the absence of any influence from mass-transfer fenomena.
In other words, the regeneration of the reducible [NiL]+ complex
(Scheme 1) occurs very fast, at the expense of
the excess free Ni2+ ion and without a noticeable concentration
gradient for this one.
The effect of temperature is also in accord with the above interpretation. The limiting current increases with the temperature according to Arrhenius equation. Such a dependence is an additional proof for the occurrence of the regeneration step as the rate determining step.
Catalytic nickel reduction by the process here described offers the possibility of determining MPR in various concentration ranges. Thus, for the 10 100 nM range, catalytic cathodic stripping voltammetry proves as being very suitable [5]. Alternatively, differential pulse polarography appears as the method of choice in the micromolar concentration range.
References (with links to reprints available in
NTNU Library, Trondheim)