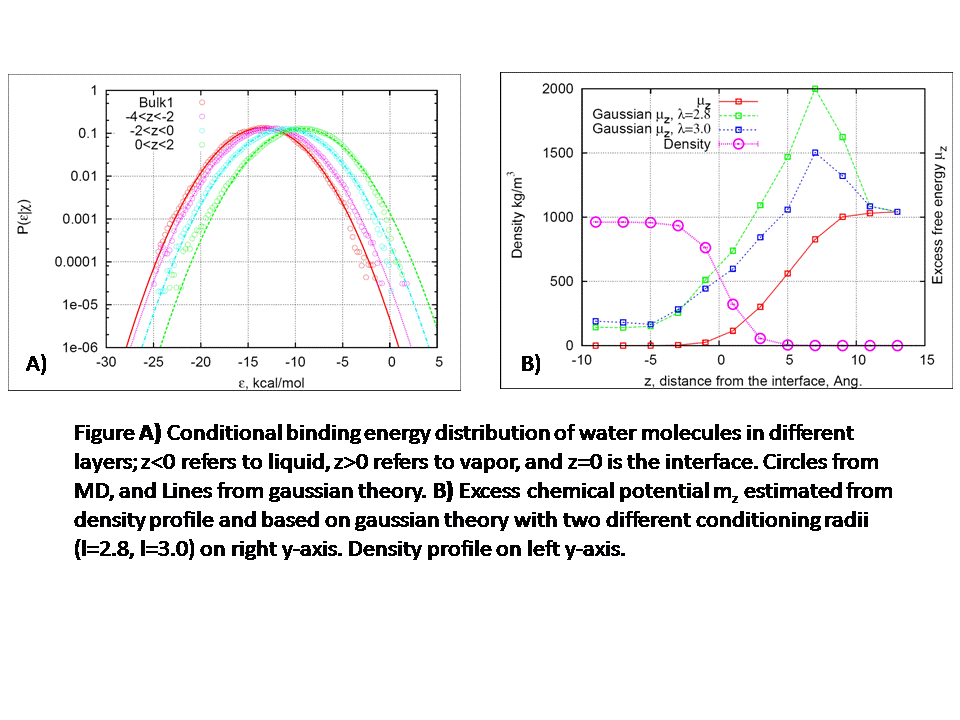
161f Quasi-Chemical Theory and Gaussian Model for Solvation at An Aqueous Interface
Recently it has been shown how quasi-chemical theory can be applied to solvation in bulk liquid phase, modeled realistically on the basis of simulation data obtained from general purpose calculations1-3. When applied to the solvation of CF4, H2O, and N(CH3)4+ in liquid water, this theory provides a concise statement of the thermodynamic balance between the short-ranged structure and long-ranged interactions. The effects of short-ranged order are captured by terms reflecting a packing problem and a chemical problem, the latter describing short-ranged attractive interactions. In the gaussian model the effects of long-ranged interactions are expressed through a conditional mean and a conditional variance. Here, the conditioning refers to collecting statistics only when the solvent molecules are at a distance larger than λ, the conditioning radius. The distribution of the binding-energy ε, (the interaction energy between solute and solvent) is denoted by P(ε|χ=1),where χ=1 refers to the case when there are no solvents within a distance of λ (otherwise χ=0). More conditioning (larger λ) amounts to more extensive numerical characterization of the short-ranged structure. With aggressive conditioning, the observed probability distributions of binding energies, P(ε|χ=1), become accurately gaussian. It is desirable to extend the quasi-chemical theory to solvation in heterogeneous environments and interfaces. The present research extends the theory to analyze simulation data on the water liquid-vapor interface, and the results are compared to the corresponding analysis of bulk liquid water. Results from an MD simulation of a TIP3P water-vapor interface is shown in Figs. A and B. Simulations of the liquid-vapor interfaces were done with slab configurations at temperatures in the range 275-350 K. We also simulated a water droplet in spherical geometry at 300 K. For analyzing the simulation snapshots, a Gibbs dividing surface was used to define the exact position of the interface. This is required to account for the capillary waves that form at the interface. The structure of the interface, orientation of the molecules, and the electrostatic potential across the interface, all agree with previous work in the published literature. In contrast to the nearly-gaussian distribution of long-ranged interactions P(ε|χ=1) in the bulk, in the interfacial region that distribution gives some indication of multi-modal behavior when only minimal statistical conditioning is imposed. Strong conditioning again achieves nearly gaussian behavior (see P(ε|χ=1) in Fig-A) but the variances observed in the interfacial region are even larger than those of the bulk liquid. For the interface problem, the observation of the interfacial density profiles provides an in situ check on the adequacy of the statistical thermodynamic theory. The excess chemical potentials (μz) calculated by the gaussian theory differs by 1-2 kcal/mol from the μz estimated from the density profile. This difference arises due the slight deviations of the binding energy distributions from the gaussian distribution, especially at high ε, as shown in Fig. A. It is still remarkable that the trends between excess chemical potential from the theory and the observations (red and blue curves Fig B) agree well in a range of ±5Ĺ from the interface. (1) Shah, J. K.; Asthagiri, D.; Pratt, L. R.; Paulaitis, M. E. J. Chem. Phys. 2007, 127, 144508 (2) Asthagiri, D.; Ashbaugh, H. S.; Piryatinski, A.; Paulaitis, M. E.; Pratt, L. R. J. Am. Chem. Soc. 2007, 129, 10133. (3) Chempath, S.; Pratt, L. R.; Asthagiri, D.; Paulaitis, M. E. “Quasi-chemical theory and the molecular binding energy distributions of water” (in preparation, 2008)