688c A New Algorithm for Spectrum Alignment by Maximizing Correlations among Signature Peaks
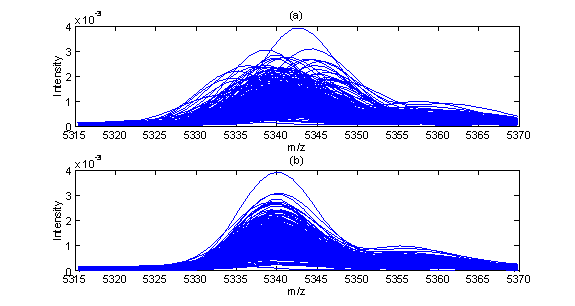
Recent advances in mass spectrometry proteomic analysis have generated considerable research interest in bioinformatics community. Large data sets generated by high-throughput mass spectrometry techniques such as SELDI-TOF (surface enhanced laser desorption/ionization time-of-flight) and MALDI-TOF (matrix assisted laser desorption and ionization time-of-flight) have been analyzed for cancer diagnostic purposes. A mass spectrometry data set contains the information about the relative abundance of many protein/peptide sequences in the form of intensity (relative abundance) measurement and molecular mass-to-charge ratio (m/z) pairs. Ideally, the same protein/peptide sequence detected in the same mass spectrometer should have the same m/z value. However, in practice this is not the case due to experimental variations. Consequently, a series of data pre-processing steps are necessary before the mass spectrometry patterns can be analyzed comparatively. Such pre-processing steps include baseline subtraction (Kohli, et al. 2005), normalization (Callister, et al. 2006, Wang, et al. 2006), peak detection, peak alignment (Nielsen et al. 1998, Bylund, et al. 2002, Tibshirani, et al. 2004, Wang, et al. 2006) and peak selection. Among all pre-processing steps, peak alignment is one of the most challenging tasks. ÝSuccessful peak alignment ensures that the same protein intensities are correctly identified in each sample spectrum to avoid errors in identifying the signals from peptides with similar molecular weight. Recent work (Semmes et al., 2005) utilizing spectrometers located in different medical centers cites difficulties with alignment/calibration as major barriers that must be overcome to ensure that data from different centers are compatible. Baggerly et al. (2004) also identify alignment problems as a significant hindrance in achieving reproducibility from samples collected within the same lab. In the past decade, several algorithms have been developed in the literature of aligning data sets, such as dynamic time warping (DTW) (Kassidas et al. 1998), derivative dynamic time warping (DDTW) and Correlation Optimised Warping (COW) (Nielsen et al. 1998). In this work, a new approach for peak detection and alignment has been developed and applied to a prostate cancer and liver cancer proteomic data sets. One difference between the proposed method and existing alignment methods is that while in existing methods all data points are aligned with equal weight, the proposed method align major peaks first, and then align other points including between-peak points and insignificant peaks based on the major peak alignment. The alignment consists of three steps. In the first step, a reference spectrum is identified and major peaks in the reference spectrum are identified automatically. In the second step, major peaks in each spectrum in the data set are aligned against the reference spectrum. Potential mismatches among major peaks are eliminated in the second step as well by automatic detection of outliers in alignment shifts. In the last step, all other points than major peaks are aligned based on the alignment results obtained in step two. The proposed method is applied to two mass spectrometry data sets. One example of alignment is shown in Fig. 1 where (a) shows before alignment; (b) shows after alignment. The performance of the proposed alignment method is compared with the classical methods DTW, DDTW and COW. The results show that the proposed method is more robust and provides better alignment.
Fig.1. A segment of sample spectra before (a) and after (b) alignment References: Baggerly,K. et al. (2004) Reproducibility of seldi-tof protein patterns in serum comparing data sets from different experiments. Bioinformatics, 20, 777ñ785. Bylund, D et al. (2002) Chromatographic alignment by warping and dynamic programming as a pre-processing tool for parafac modelling of liquid chromatography-mass spectrometry data. J. Chromatography, 961:237-244. Callister, SJ et al.(2006) Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res., 5:277-86. Kassidas, A, MacGregor, JF, Kohli, BM et al. (2005) An alternative sampling algorithm for use in liquid chromatography/ tandem mass spectrometry experiments. Rapid Commun. Mass Spectrometry, 19(5):589-596. Nielsen, NPV, Carstensen, JM, Smedsgaard, J. (1998) Aligning of single and multiple wavelength chromatographic profiles for chemometric data analysis using correlation optimised warping. J. Chromatogr. A; 805: 17ñ35. Tibshirani, R et al. (2004) Sample classi_cation from protein mass spectrometry, by 'peak probability contrasts'. Bioinformatics, 20(17):3034-44. Semmes,O. et al. (2005) Evaluation of serum protein profiling by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry for the detection of prostate cancer: Wang, P et al. (2006) A statistical method for chromatographic alignment of LC-MS data. Biostatistics, doi:10.1093/biostatistics/kxl015.